Ab-initio simulace funkčních materiálů pro zelené technologie
NFPL220
doc. RNDr. Vojtěch Chlan, Ph.D.
RNDr. Jan Kuriplach, CSc.
přednáška:
- pondělí 15:10 - 16:40 učebna T9
- středa 14:50 - 16:20 učebna C126KFNT
-
1. přednáška: Pásová struktura, translační symetrie, reciproká mřížka, vlnová funkce, elektronová struktura
-
2. přednáška: Bornova-Oppenheimerova aproximace, Hartreeho-Fockova metoda, Teorie funkcionálu hustoty, Kohnovi-Shamovy rovnice, Metody výpočtu elektronové struktury, metoda LAPW
-
3. přednáška: Rychlý úvod do HPC computing, linux,
1. cvičení: cviceni0102_initWIEN2k.pdf WIEN2K: získání a sestavení vstupní struktury
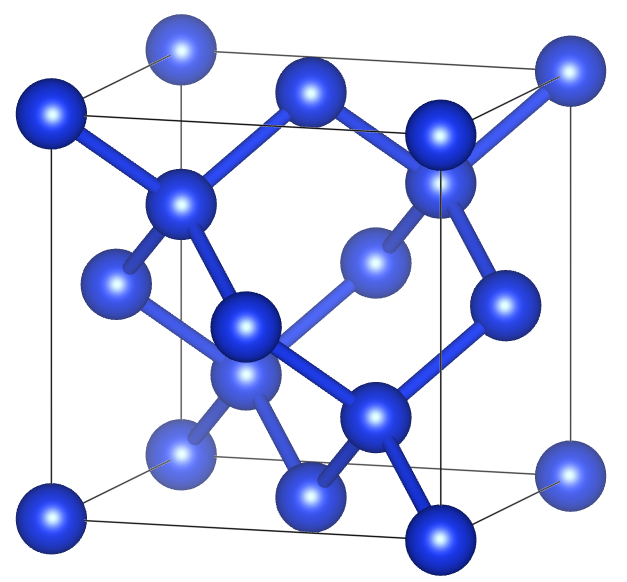
- diamantová struktura křemíku - získání:
- Použijeme CIF nalezený v Open Crystallography Database: 9008565.cif
- WIEN2k pro prostorovou grupu #227 (Fd-3m) podporuje setting 2 (ale CIF z OCD má setting 1)
- Před konverzí pomocí cif2struct je tedy potřeba změnit:
název grupy z 'F d -3 m :1' na 'F d -3 m'
souřadnice Si atomu ze 0.00000 0.00000 0.00000 na 0.12500 0.12500 0.12500 - Výsledný cif již lze zkonvergovat: cif2struct diamSi.cif → diamSi.struct
- Je vhodné dát nějaký title na první řádek a zkontrolovat programem sgroup:
x sgroup → diamSi.struct_sgroup
- nástroj na zobrazování struktur se spoustou užitečných funkcí: VESTA
- Cvičení 1
-
2. cvičení: cviceni0102_initWIEN2k.pdf
WIEN2K: inicializace výpočtu - technické parametry
Použijeme interaktivní skript init_lapw, je toho hodně, ale vlastně skoro všechny naše vstupy budou "defaultní" hodnoty.
- setrmt se zeptá na procentuální zmenšení všech atomových koulí (RMT). 0 % znamená téměř dotýkající se koule.
7 <ENTER> (chceme trochu prostoru pro objemovou optimalizaci struktury, RMT Si by nově mělo být 2.05 a.u.) - setrmt: Use old or new scheme (o/N)
<ENTER> (default, nové schéma nastavuje vhodné proporce mezi RMT různých prvků - zde se neuplatní)
a <ENTER> (přijmout změnu RMT vyrobenou programem setrmt)
- setrmt se zeptá na procentuální zmenšení všech atomových koulí (RMT). 0 % znamená téměř dotýkající se koule.
- nn: specify nn-bondlength factor: (usually=2)
2 <ENTER> (default)
V automaticky otevřeném diamSi.outputnn si můžeme zkontrolovat, že každý Si má 4 nejbližší Si sousedy ve vzdálenosti 2.35156 Å.
Soubor diamSi.outputnn zavřeme (:q) a pokračujeme programem sgroup: c <ENTER> - sgroup: kontrola struktury a případné navržení nové. Zde sgroup navrhne pouze (zbytečné) prohození os, což není nutné přijmout.
V automaticky otevřeném diamSi.outputsgroup si můžeme vše prohlédnout, např. že prostorová grupa je: 227 (F d -3 m) [origin choice 2].
Soubor diamSi.outputsgroup zavřeme (:q) a pokračujeme programem symmetry: c <ENTER>
(Kdybychom navrženou změnu struktury chtěli přijmout, dáme volbu: e <ENTER>.) - symmetry: kontrola struktury, pro každý atom nalezení bodové grupy a nastavení takového lokálního systému, aby byla použitá sférická báze (Ylm) co nejúspornější.
V automaticky otevřeném diamSi.outputs si můžeme vše prohlédnout, např. že bodová grupa Si je -43m a lokální systém nebylo potřeba rotovat (LOCAL ROT MATRIX je jednotková matice).
Soubor diamSi.outputs zavřeme (:q) a pokračujeme programem lstart: c <ENTER> - init_lapw nejprve spustí instgen, který pomůže s nastavením spinové konfigurace.
My chceme Si uvažovat nemagnetický, takže: -nm <ENTER> - lstart: jako výměnně-korelační potenciál budeme používat GGA-PBE.
13 <ENTER> (default)
Zvolíme hranici pro odlišení core a valence stavů:
-6 <ENTER> (default)
V otevřeném diamSi.outputst si můžeme prohlédnout, jak lstart postupoval, a zavřít ho (:q). Rozdělení dopadlo pro -6 Ry takto:Atomic configuration for atom: Si Z= 14.00 E-up(Ry) E-dn(Ry) Occupancy q/sphere core-state 1S -131.253564 -131.253564 1.00 1.00 1.0000 T 2S -10.251641 -10.251641 1.00 1.00 0.9999 T 2P* -7.053686 -7.053686 1.00 1.00 0.9996 T 2P -7.006118 -7.006118 2.00 2.00 0.9996 T 3S -0.794415 -0.794415 1.00 1.00 0.5027 F 3P* -0.301274 -0.301274 0.50 0.50 0.3006 F 3P -0.298957 -0.298957 0.50 0.50 0.2979 FNáš Si bude mít 10 elektronů (1s, 2s, a 2p stavy) jako core, zbylé 4 elektrony budou uvažovány jako valenční.
Tímto způsobem se nakonfigurovaly oba Si atomy ve struktuře, můžeme pokračovat: c <ENTER>
(Kdybychom něco v této konfiguraci chtěli změnit, zrestartujeme lstart volbou: e <ENTER>.) - init_lapw otevře diamSi.in1, kde můžeme měnit parametry (budoucí) báze. Nejdůležitější je R-MT*K-MAX na druhém řádku.
Ponecháme defaultní 7.0 a pokračujeme (:q) - init_lapw otevře diamSi.in2, kde zvýšíme parametr GMAX na předposledním řádku z 12.0 na 14.0 a soubor uložíme (:wq!).
- kgen: jako počet k-bodů v celé Brillouinově zóně zvolíme: 200 <ENTER>.
Otevře se soubor diamSi.klist, kde si můžeme zkontrolovat, že v ireducibilní části BZ tomu odpovídá 10 k-bodů.
Soubor zavřeme (:q) a pokračujeme programem dstart: c <ENTER> - dstart provede superpozici atomových hustot obou Si (z lstart) a finální hustotu vyjádří už v bázi, která se bude ve výpočtu používat, tj. podle námi definovaných parametrů (RKmax,Gmax, atd.).
Otevře se soubor diamSi.outputd, kde si můžeme leccos zkontrolovat. (:q) - init_lapw vygeneruje zbývající vstupní soubory a zeptá se, zda chceme spinově polarizovaný výpočet.
To pro nemagnetický Si nechceme: n <ENTER>
A tím je inicializace dokončena, výpočet (SCF) lze spustit.
-
3. cvičení: cviceni03_WIEN2K_spusteni.pdf
WIEN2K: spuštění výpočtu, čtení a filtrování výsledků
-
4. cvičení: cviceni04_WIEN2k_optimalizace.pdf
WIEN2K: optimalizace struktury, DOS, bandstructure, struktura Si (diamantová vs. beta-Sn)
Zístkání některých dalších vlastností, které nejsou přímo spočteny během SCF.
(Obvykle použijeme finální, zoptimalizovanou strukturu.)
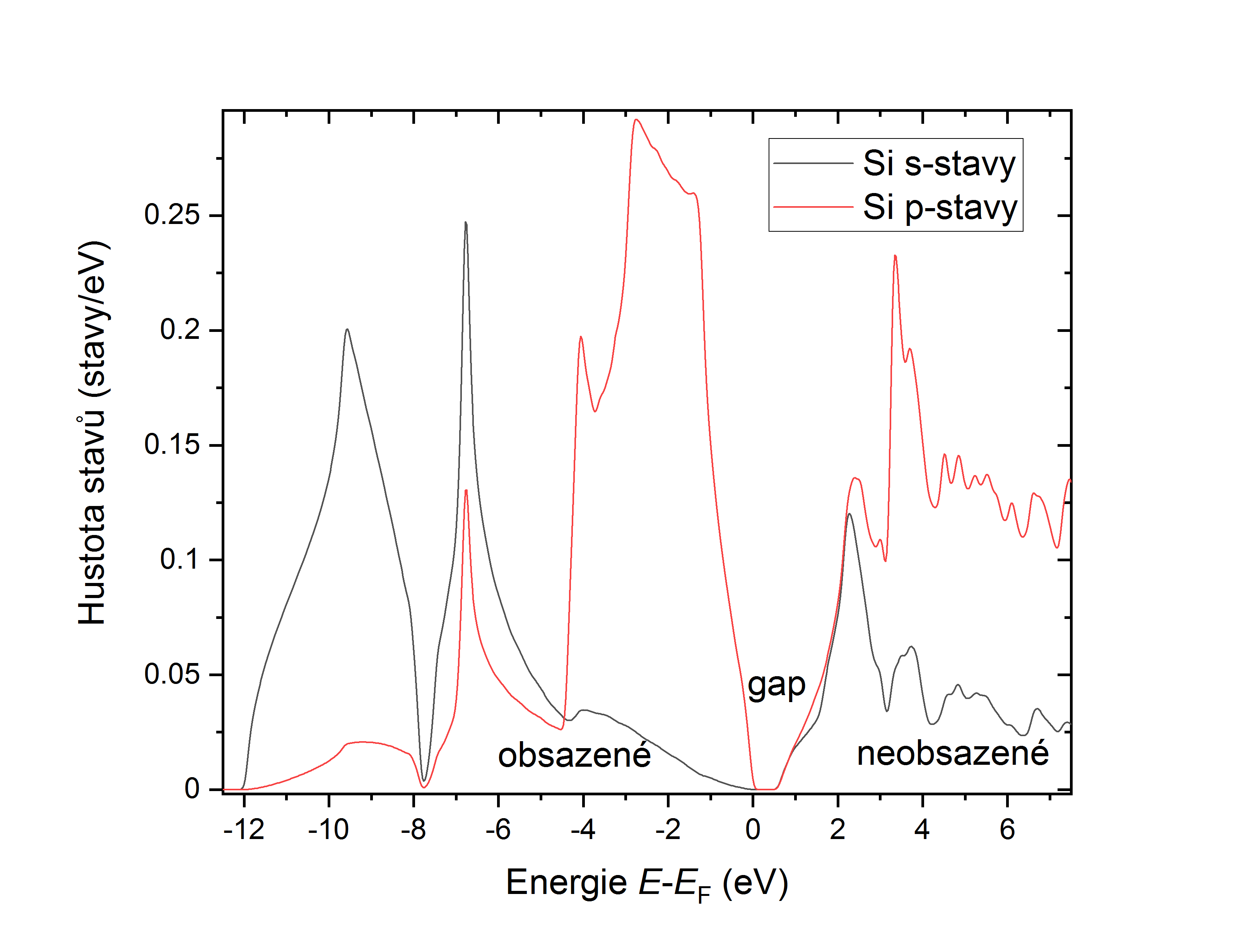
Spočtení hustoty stavů (DOS)
- x kgen: použijeme hustší k-mesh, např. 10000 k-bodů v BZ
To vygeneruje 256 k-bodů (21x21x21) v ireducibilní části BZ.
(Srovn. s 10 k-body a dělením 5x5x5 postačujícími pro normální výpočet.) - x lapw0: znovu vygenerujeme potenciál
- x lapw1: vyřešíme eigenproblém na novém, hustším k-meshi
- x lapw2 -qtl: spočítáme náboj (pro dané L a M) pro všechny atomy (uvnitř RMT).
Výsledek se zapíše do diamSi.qtl. - Vyrobíme vstupní soubor diamSi.int pro program tetra (např. úpravou SRC_templates/case.int):
- x tetra: spočítáme DOS, výsledek je ve sloupcích v diamSi.dos (v Ry) a diamSi.dos1ev (v eV, Fermiho hladina = 0 eV).
DOS si pak lze nakreslit ve vhodném progamu.
Title -9.50 0.002 1.500 0.003 # EMIN, DE, EMAX, Gauss-broadening(>de) 2 N 0.000 # NUMBER OF DOS-CASES below, G/L/B broadening (Ry) 1 2 total # atom, case=column in qtl-header, label 1 3 total # atom, case=column in qtl-header, labelČísla pro ojednotlivé orbitaly a suborbitaly zde odpovídají legendě v diamSi.qtl, která je v našem případě tot,0,1,2,D-eg,D-t2g,3.
Takže řádky s volbami 2 a 3 v diamSi.int vykreslí pouze hustotu pro s a p stavy uvnitř atomové koule Si.
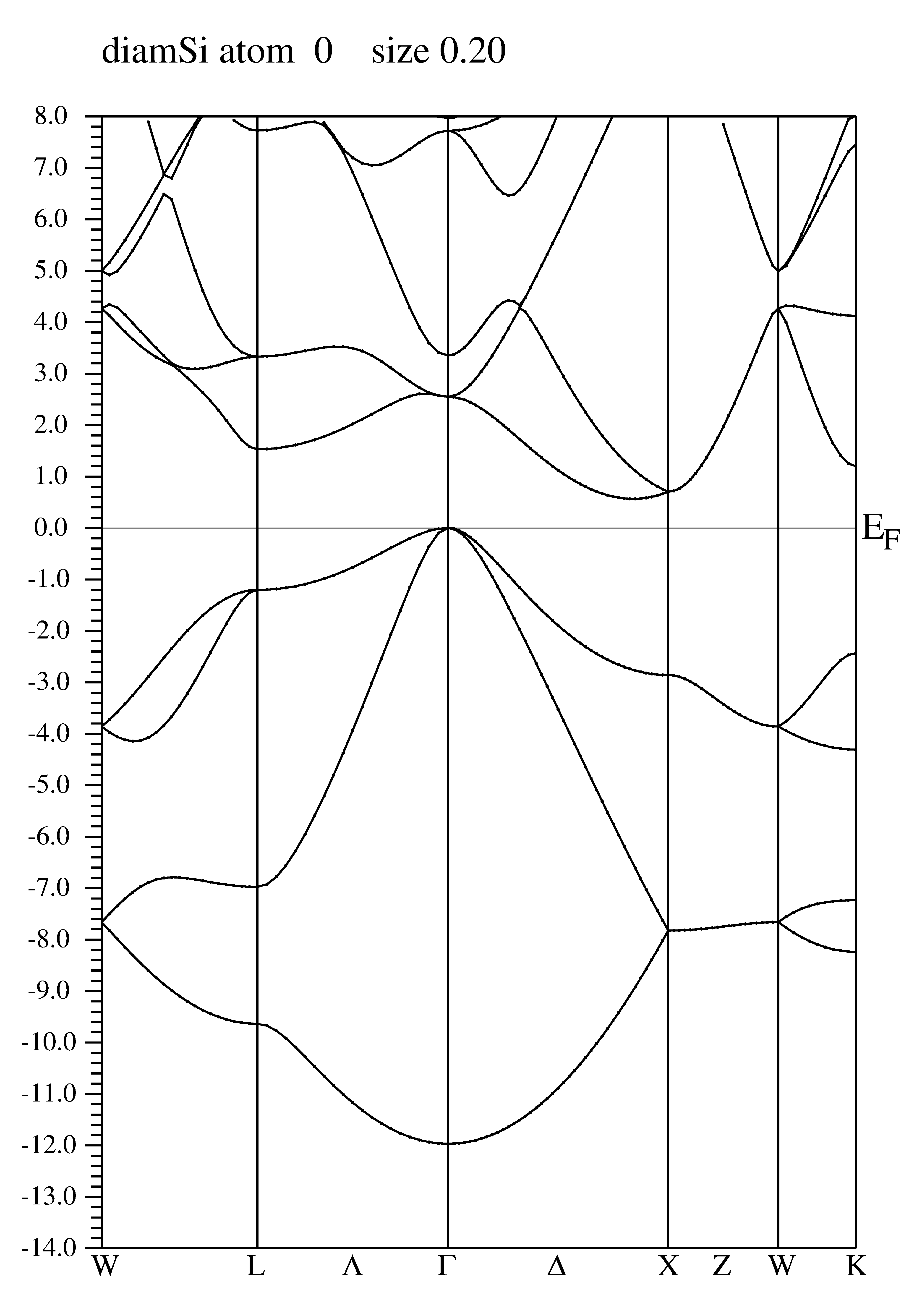
Spočtení pásové struktury (bandstructure)
- Definujeme speciální body v k-prostoru, obvykle tak aby tvořily trajektorii procházející významné body symetrie.
Pro diamantovou strukturu Si lze použít trajektorii W-L-Λ-Γ-Δ-X-Z-W-K v SRC_templates/fcc.klist nebo její část (případně jakkoliv upravit). - Výslednou trajektorii uložíme do diamSi.klist_band .
- x lapw0: znovu vygenerujeme potenciál
- x lapw1 -band: vyřešíme eigenproblém na této trajektorii (k-path)
- x lapw2 -band: spočítáme energie pro body na naší trajektorii v k-prostoru.
(Lze zkombinovat s -qtl volbou (viz DOS) pro zdůraznění orbitálního charakteru vykreslených pásů.) - Vyrobíme vstupní soubor diamSi.insp pro program spaghetti (např. úpravou SRC_templates/case.insp):
- V diamSi.insp si lze nastavit vhodné rozměry a další vlastnosti výstupu
- x spaghetti: spočítáme pásovou strukturu, výsledek je postscriptový obrázek diamSi.spaghetti_ps :
### Figure configuration 2.0 2.0 # paper offset of plot 14.0 21.0 # xsize,ysize [cm] 1.0 4 # major ticks, minor ticks 1.0 1 # character height, font switch 1.0 3 3 # line width, line switch, color switch ### Data configuration -14.0 8.0 2 # energy range, energy switch (1:Ry, 2:eV) 1 0.3946231 # Fermi switch, Fermi-level (in Ry units) 1 999 # number of bands for heavier plotting 1,1 0 1 0.2 # jatom, jcol, size of heavier plotting
- x kgen: použijeme hustší k-mesh, např. 10000 k-bodů v BZ
- 5. přednáška: prednáška 5 Symetrie krystalové mřížky v materiálech, Pseudopotenciál, úvod do VASP
- 5. cvičení:
- cviceni04_VASP_input.pdf VASP: FeAl příklad
- 6. přednáška: prednáška 6 kmity atomův materiálech, fonony
- 6. cvičení: cviceni04_VASP_input.pdfLiFePO4
Další studijní materiály:
-
Úvod do výpočtů elektronové struktury
- základní principy, vztah k fyzice pevných látek a materiálům
- Pásová elektronová struktura pevných látek
- Teorie funkcionálu hustoty, Kohnovy-Shamovy rovnice a základní metody jejich řešení
- Výměna a korelace v elektronovém plynu
-
Úvod do počítání ve VASP a WIEN2k, úvod do počítání na HPC
- Metody výpočtu elektronové struktury
- Ab-initio molekulární dynamika
-
Základní příklady (tutorial)
-
Modelování pokročilejších specifických příkladů
- materiály pro absorpci a skladování vodíku
- materiály pro baterie
- materiál pro fotovoltaiku
- porézní materiály pro skladování, separaci, a katalýzu plynů a jiných látek
-
Studijní literatura:
- Kieron Burke, The ABC of DFT (dft.uci.edu/doc/g1.pdf)
- P. Blaha, K.Schwarz, F. Tran, R. Laskowski, G.K.H. Madsen and L.D. Marks, J. Chem. Phys. 152, 074101 (2020)
- S. Cottenier, Density Functional Theory and the family of (L)APW-methods: a step-by-step introduction, 2002-2013 (2nd edition), ISBN 978-90-807215-1-7,
- The VASP Manual, www.vasp.at/wiki/index.php/The_VASP_Manual